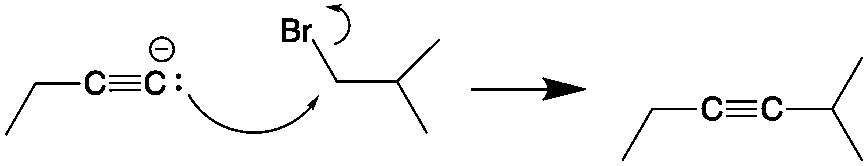
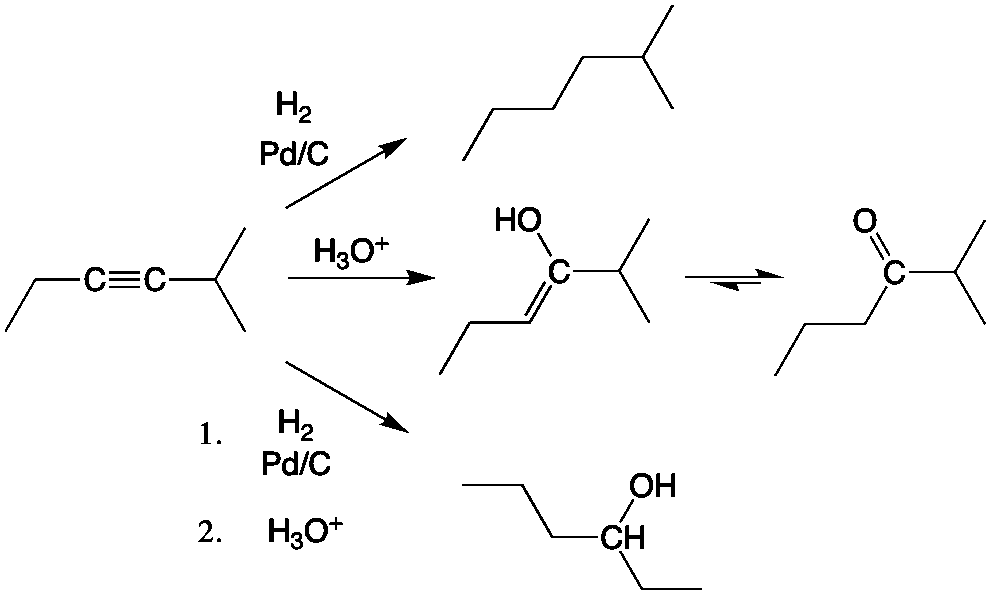
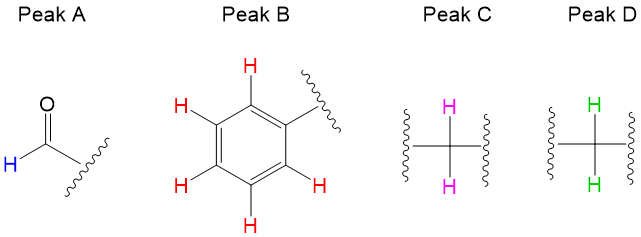
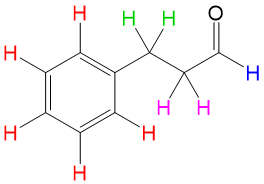
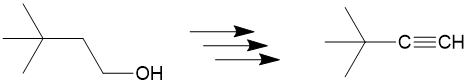
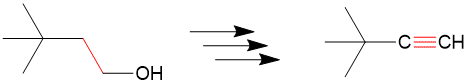
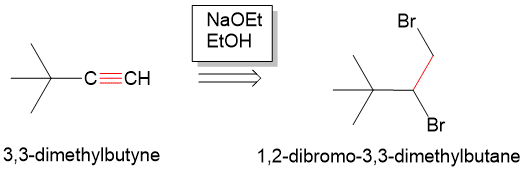
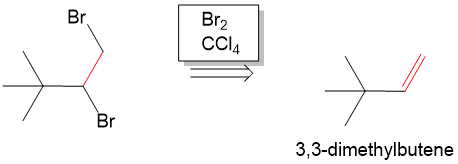
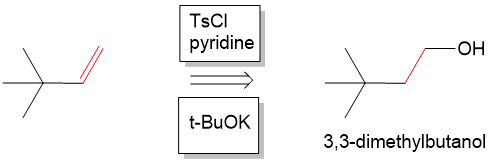
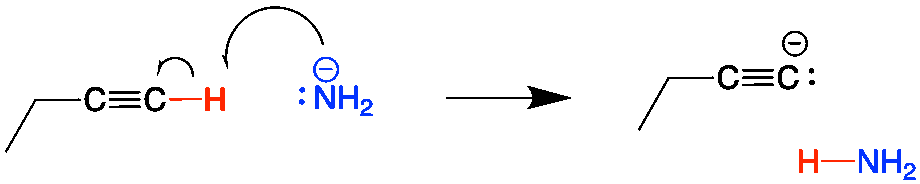In addition to organic chemistry, understanding human anatomy helps connect scientific principles to real-world biological function. The human body is a complex system where structure and function are closely linked, and one of the most interesting examples of this relationship is the nose.
The structure of the nose plays a key role in both breathing and appearance, which is why procedures like nose reshaping surgery are sometimes used to improve function and balance.
While often associated with appearance, the nose plays a vital role in respiratory health, air filtration, and overall facial balance. Studying nasal anatomy provides insight into how even small structural variations can impact both function and symmetry.
The Structure of the Nose
The nose is composed of both bone and cartilage, forming a framework that supports its shape and function. The upper portion consists primarily of nasal bones, while the lower portion is made up of flexible cartilage that allows for movement and structural variation.
Inside the nose, the nasal septum divides the cavity into two passages. This structure plays a key role in directing airflow. When the septum is straight, air can move efficiently through both sides. However, when it is deviated or irregular, it can restrict airflow and lead to breathing challenges.
Function: More Than Just Breathing
As air enters the nasal passages, it undergoes several important processes. The nose filters out dust, allergens, and other particles, while also humidifying and warming the air before it reaches the lungs. This helps protect the respiratory system and maintain optimal function.
Specialized structures within the nose, such as the turbinates, increase surface area and improve the efficiency of this process. These features demonstrate how anatomical design directly supports physiological function.
Nasal Anatomy and Facial Balance
Beyond its functional role, the nose is a central feature of the face and significantly influences overall appearance. Its size, shape, and projection affect how other facial features—such as the chin, jawline, and forehead—are perceived in relation to one another.
In studies of facial proportions, balance and symmetry are often linked to how well these features align. Even subtle changes in nasal structure can alter the visual harmony of the face, which is why the nose is often a focal point in both scientific and medical discussions of facial anatomy.
When Structure and Function Intersect
In some cases, individuals may experience structural concerns that affect both breathing and appearance. These may result from natural development, injury, or anatomical irregularities such as a deviated septum.
When these issues extend beyond what can be addressed through non-invasive methods, medical procedures such as rhinoplasty may be considered. From a scientific perspective, this procedure focuses on modifying nasal structure to improve airflow, restore balance, and support both functional and aesthetic outcomes when performed by a qualified specialist.
The Importance of Studying Anatomy
Exploring topics like nasal anatomy highlights the importance of understanding how different systems in the body are interconnected. It reinforces the idea that form and function are not separate concepts, but rather deeply integrated components of human biology.
For students studying science, medicine, or health-related fields, this serves as a practical example of how theoretical knowledge can be applied to real-world scenarios. By understanding structure at both the macro and micro levels, we gain a deeper appreciation for how the body operates as a whole.
Conclusion
The nose is far more than a defining facial feature—it is a sophisticated structure that plays a critical role in both physiology and appearance. Through the study of anatomy, we can better understand how structural variations influence function, and how science continues to inform advancements in medical and surgical techniques.