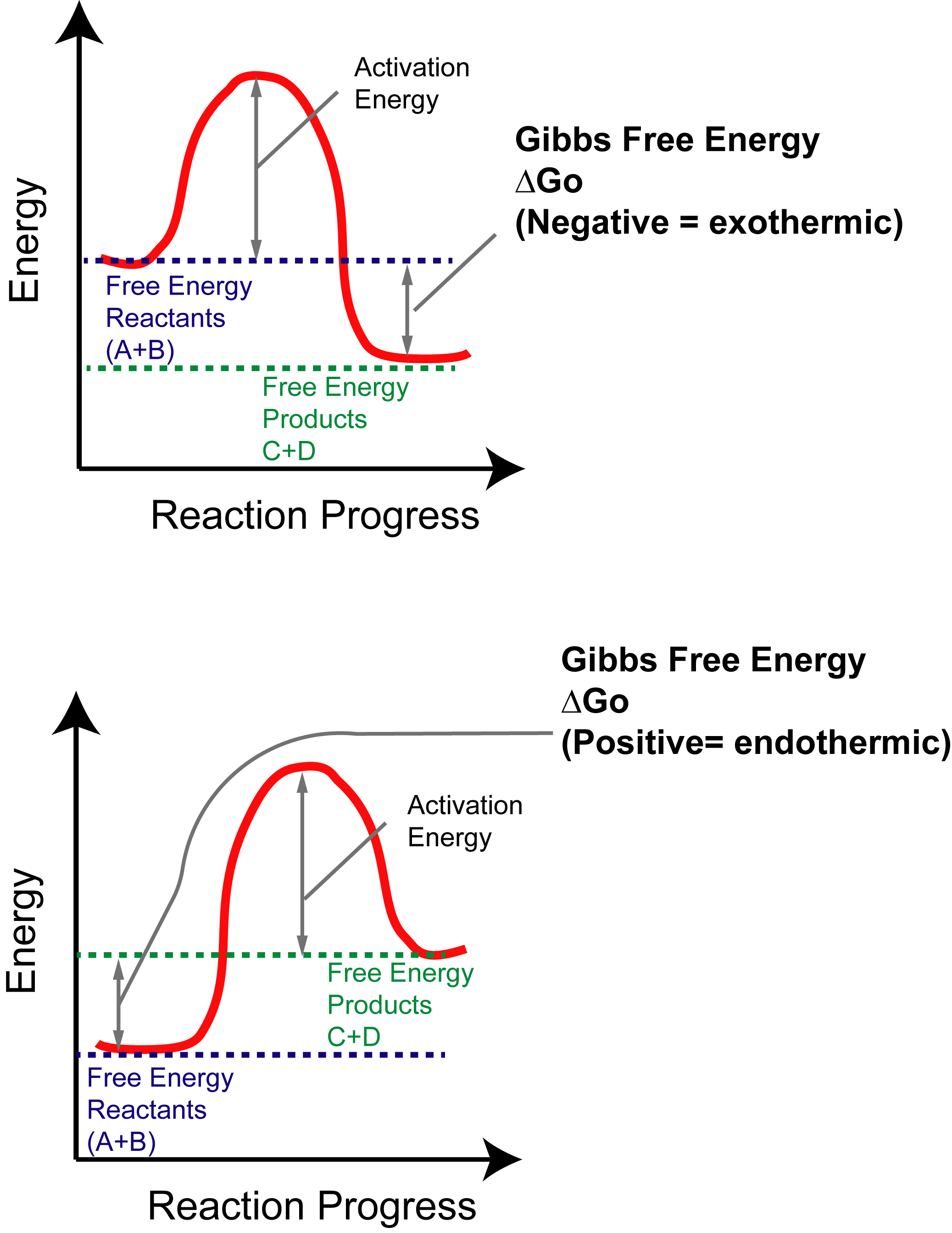
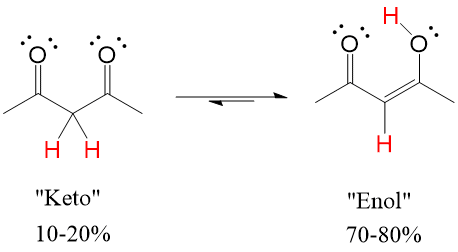
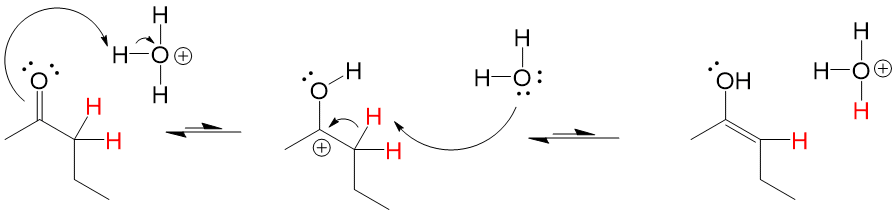
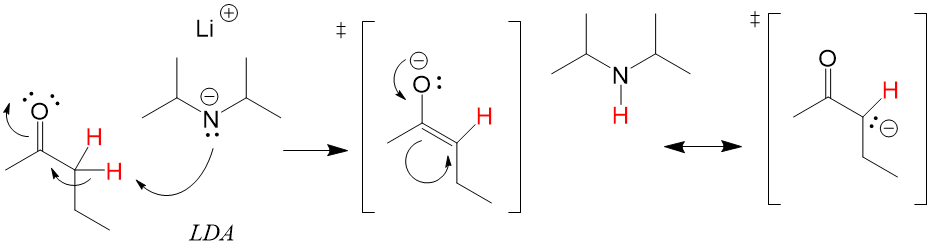
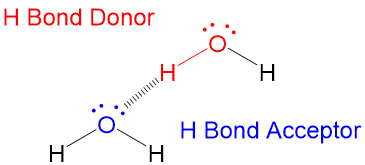
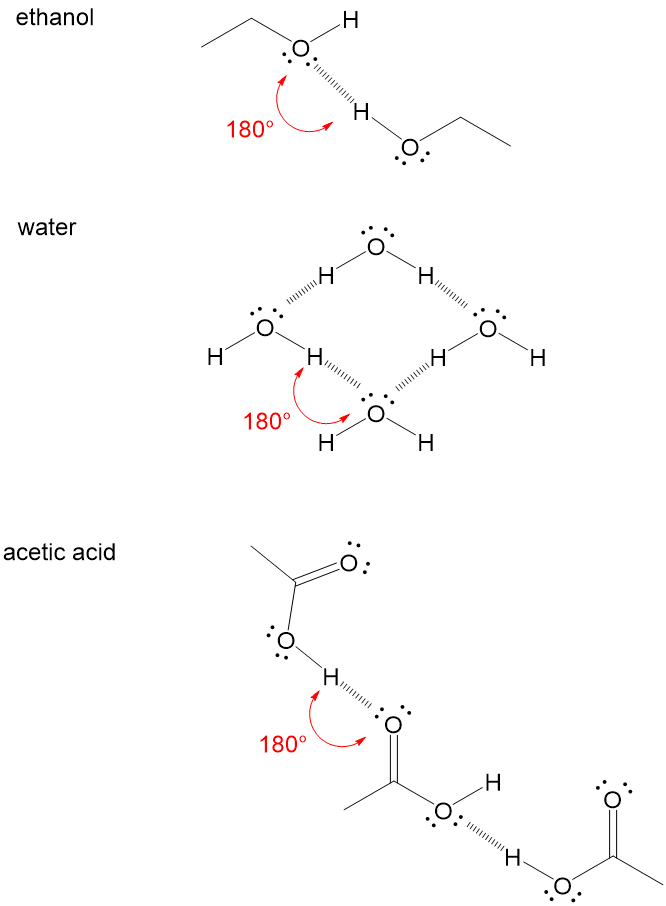
One of the difficult things to learn in Orgo1 is 3D drawing on 2D paper. This is as much a skill of art as science and can be very difficult for students to pick up, making the process harder to learn. Here at StudyOrgo.com, we have come up with clear-cut explanations and descriptions of hard-to-learn concepts and organized them on our website so you can spend more time practicing and less time searching.
Cycloalkanes can be one of the hardest things to draw in this class. First we will address the Haworth projection,
What is the chair conformation?
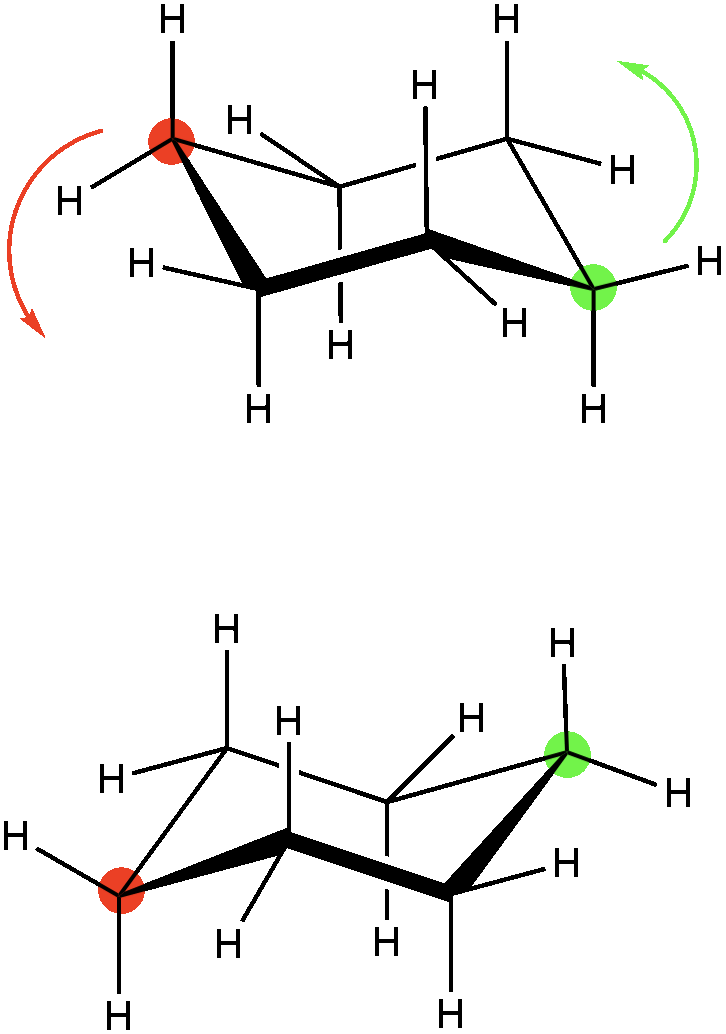 Because there are six sp3 hybridized carbons in a cyclohexane ring, the chair conformation becomes the most stable conformation. The ring can be twisted in several orientations that are all higher in energy and thus less stable, but because the ring can be twisted it can potentially become inverted, which would flip all of the substituents of the ring in the opposite order. In the example below, if we twisted the red and green carbon in the direction of the arrows, the ring would become “flipped” have an equally stable conformation. In solution, both are possible unless there is steric strain that prevents one conformation over the other.
Because there are six sp3 hybridized carbons in a cyclohexane ring, the chair conformation becomes the most stable conformation. The ring can be twisted in several orientations that are all higher in energy and thus less stable, but because the ring can be twisted it can potentially become inverted, which would flip all of the substituents of the ring in the opposite order. In the example below, if we twisted the red and green carbon in the direction of the arrows, the ring would become “flipped” have an equally stable conformation. In solution, both are possible unless there is steric strain that prevents one conformation over the other.
How do I draw a chair conformation correctly?

First, draw the structure VERY BIG. This will make it much more easy to draw in the substituents. To draw this projection, 4 steps are needed:
Step 1: Draw a very flat and wide “V”.
Step 2: Draw two lines at an angle away from the “V”.
Step 3: Draw one line that comes up at a steeper angle than the “V” and then shorter and off of the center of the “V” (i.e. sorter than where the dotted line is).
Step 4: Draw the last line that closes the ring.
What is Axial and Equatorial?
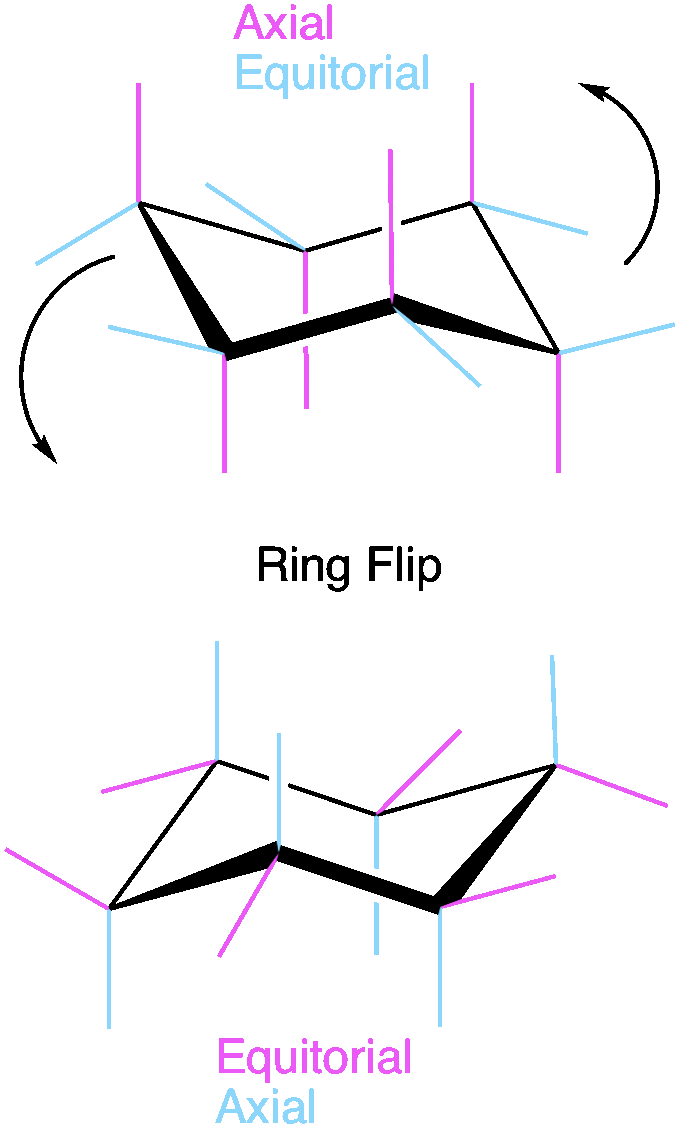 Because two bonds to each carbon are required to from the cyclohexane ring, each carbon can have two “substituents” or R groups. One will be parallel to the ring in the “equatorial position” the other will be orthogonal to the ring and will be in the “axial” position. Let’s look at his example. If we start with pink bonds in axial and blue bonds in equatorial, a ring flip will invert this and place the blue bonds in axial and pink bonds in equatorial. Both exist in solution UNLESS there is steric hindrance present that makes one conformation less stable. Any bulky group or electronegative group will prefer to be in the equatorial position. This is because the equatorial position places the substituent away from the ring where there is free space for the substituent. On the other hand, bulky groups in the axial position will experience “clashing” with the other axial positions because space is limited. The more substituents on the ring, the more important it is to consider this when drawing the “most stable” conformation.
Because two bonds to each carbon are required to from the cyclohexane ring, each carbon can have two “substituents” or R groups. One will be parallel to the ring in the “equatorial position” the other will be orthogonal to the ring and will be in the “axial” position. Let’s look at his example. If we start with pink bonds in axial and blue bonds in equatorial, a ring flip will invert this and place the blue bonds in axial and pink bonds in equatorial. Both exist in solution UNLESS there is steric hindrance present that makes one conformation less stable. Any bulky group or electronegative group will prefer to be in the equatorial position. This is because the equatorial position places the substituent away from the ring where there is free space for the substituent. On the other hand, bulky groups in the axial position will experience “clashing” with the other axial positions because space is limited. The more substituents on the ring, the more important it is to consider this when drawing the “most stable” conformation.
What is Diaxial Strain?
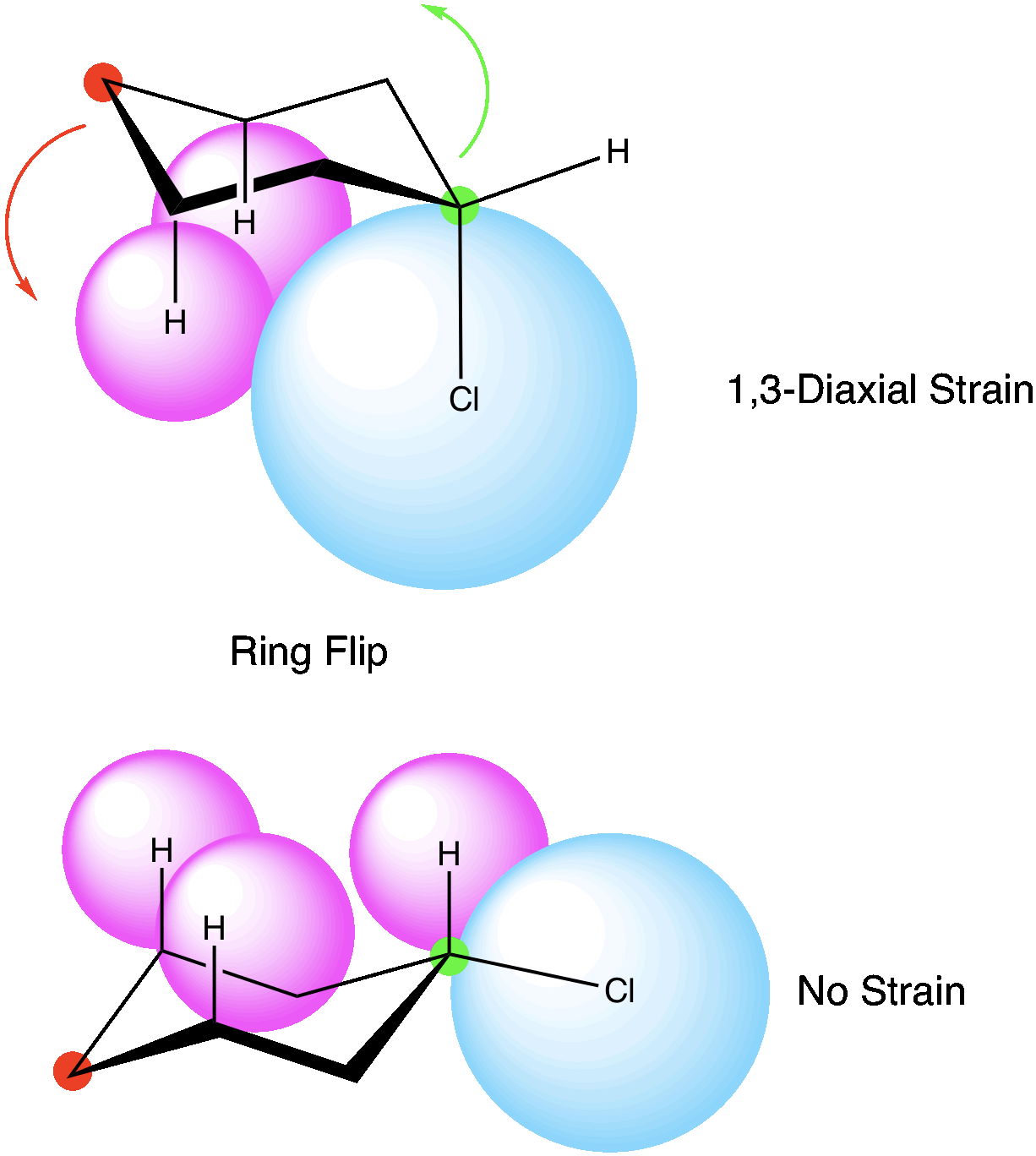 Let’s look at chlorocyclohexane, there are two possible conformations. In one case, the chlorine atom is in the axial position and is experiencing 1,3 diaxial strain with the other hydrogens in the axial position. If we do a ring flip, we place the chlorine now in the equatorial position, and only hydrogens are in the axial position. This would be the most stable conformation.
Let’s look at chlorocyclohexane, there are two possible conformations. In one case, the chlorine atom is in the axial position and is experiencing 1,3 diaxial strain with the other hydrogens in the axial position. If we do a ring flip, we place the chlorine now in the equatorial position, and only hydrogens are in the axial position. This would be the most stable conformation.
Sign up with StudyOrgo today to get access over 180 chemical to reactions with detailed reaction mechanisms, explanations, tips and tricks to each mechanisms!